
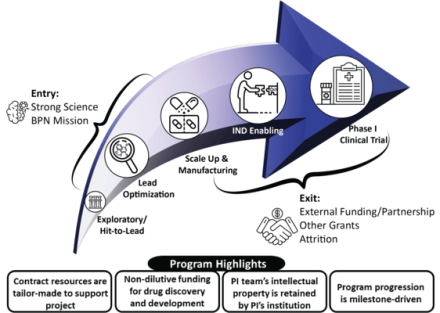
The NIH Blueprint Neurotherapeutics Network (BPN) supports drug development for nervous system disorders by providing non-dilutive funding for small molecule discoveries from hit-to-lead chemistry to phase I clinical testing. The BPN aims to transform basic research into clinical candidates and generate data to attract further funding or partnerships.
- PAR-25-051: Blueprint Neurotherapeutics Network (BPN): Small Molecule Drug Discovery and Development of Disorders of the Nervous System (UG3/UH3 Clinical Trial Optional)
Scientific Project Managers
Pascal Laeng, PhD, Point of Contact
Oreisa O'Neil-Mathurin, MPH, External Oversight Committee (EOC) Liaison
Ranga Rangarajan, PhD, Subject Matter Expert (SME) Liaison
BPN Frequently Asked Questions (FAQs)
What does a target product profile look like?
A Target Product Profile (TPP) is a planning tool for therapeutic candidates based on FDA Guidance for Industry and Review Staff Target Product Profile — A Strategic Development Process Tool(pdf, 38 KB). Download the Target Product Profile Worksheet(pdf, 38 KB) based on the FDA guidance that defines the minimal/ideal profile of the final marketed product and shows the ultimate goals of the proposed therapy development effort.
How do I develop milestones for my project?
- Provide sufficient details on methods, assumptions, experimental designs, data analysis plan and specify the quantitative criteria for measuring success and related rationale.
- Quantitative criteria should be robust and be consistent with the state-of-the-art in the field. Most of the time, the quantitative criteria for success in the milestones will also be used for making go/no-go decisions and this should be specified. If a criterion is not to be used for making go/no-go decisions, it should be clearly noted and justified.
- Specify the timeline for each milestone.
- Be aggressive yet realistic
- Define at least one milestone for each year of funding
View the BPN milestone orientation(pdf, 314 KB) for more information
Contract Research Organizations (CROs)
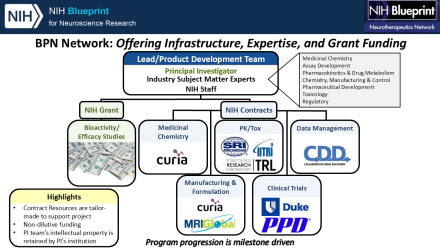
BPN researchers receive no-cost access to the network's Contract Research Organizations (CROs). LDTs plan and coordinate studies conducted by BPN CROs.
Subject Matter Experts (SMEs)
Each Lead Development Team (LDT) receives support from BPN subject matter expert. SMEs are assigned to a project with considerations for the project team's individual needs and stage of entry. LDTs are assembled to support each investigator-driven project and include the investigator’s research and development team, NIH Program staff, NIH-contracted drug development subject matter experts, and CROs. Bi-weekly team meetings and frequent email communication facilitate study planning and coordination, troubleshooting, and achieving project milestones.
Gian Luca Araldi, Ph.D., Pharm.D., M.B.A., Pharmaceutical development
Dr. Araldi is a pharmaceutical and biotechnology executive with more than 25 years of experience having responsibilities ranging from basic research through clinical development at major pharmaceutical companies including Glaxo, Forest Laboratories and Merck-Serono. Dr. Araldi has been involved in the development of key medicines such as Teflaro®, Dutogliptin®, Gavestinel®, Sanfetrinem® and Sanfetrinem® Cilexetil and is inventor of 113 granted patents. His areas of expertise span from medicinal chemistry to process development, manufacturing, and project management. Throughout his career, he has worked in diverse therapeutic areas which include oncology, cardiovascular disease, neurological disorders and immunology where he has obtained many breakthrough results. He has fostered constructive relationships with many academic labs, biotech, pharma companies and government institutions both domestic and foreign. Dr. Araldi has a doctorate degree in Organic Chemistry and a doctorate degree in Pharmacy awarded both from the University of Parma (Italy) and recently received his M.B.A. in Finance from Long Island University Post and Project Management certification from Stony Brook University.
Marc Bailie, D.V.M., Ph.D., Toxicology
Dr. Bailie is the Chief Development Officer at Integrated Nonclinical Development Solutions Inc., a drug development consulting company, and the Director of the In Vivo Facility in the Department of Pharmacology and Toxicology at Michigan State University. Prior to his current roles, he spent 11 years at Parke-Davis/Pfizer, building and directing the Safety Pharmacology Group within Drug Safety Research and Development. Dr. Bailie also has extensive experience in a broad array of animal models for the evaluation of pharmacologic and toxicologic activity of compounds. Over his tenure at Parke-Davis/Pfizer, he served on numerous Discovery and Development project teams across a variety of therapeutic areas, provided scientific direction for safety pharmacology both locally and on a global basis, and served as expert council for cardiovascular and general safety pharmacology within and outside of Pfizer. Dr. Bailie has published over 30 papers in peer-reviewed journals, and has led or participated in several expert working groups sponsored by the ISLI Health and Environmental Sciences Institute.
Phil Carpino, Ph.D., Medicinal Chemistry
Dr. Carpino has worked for over 30 years in the pharmaceutical industry as a medicinal and synthetic organic chemist, with a focus on small molecule drug discovery. He has held senior scientific research positions at Pfizer and Janssen as well as leadership positions at several biotech companies, including at X-Chem where he was an executive director in their medicinal chemistry services group. His experience spans all stages of small molecule drug discovery, from target identification/validation and high throughput screen triage to lead optimization and clinical candidate delivery. He has worked on the discovery of numerous drugs that acted at molecular targets located in the central nervous system, mostly anti-obesity targets such as NPY Y1R antagonists, CB1R antagonists, MC4R agonists, and ghrelin receptor antagonists. He has contributed to the discovery of 12 clinical candidates, 9 of which reached Phase I clinical trials and 3 of which reached Phase II clinical trials. He is a co-inventor of capromorelin, a ghrelin receptor agonist which received FDA approval for the treatment of weight loss in companion animals. He is widely recognized for his work in uncovering a covalent mechanism of action for a novel series of small molecule GLP-1R agonists/positive allosteric modulators, the discovery of which led to development of a novel sensitized high-throughput screening assay and eventually to the identification of the first potent small molecule GLP-1R agonists to progress into human clinical trials. Dr. Carpino earned his Ph.D. at Harvard University and is a co-inventor/author on 43 issued US patents and 56 scientific articles.
Nicholas Carruthers, Ph.D., Medicinal Chemistry
Dr. Carruthers is a medicinal chemist with over 35 years broad, R&D specific, executive experience within the pharmaceutical industry. He has expertise in drug discovery across several therapeutic areas, leading to multiple drug candidates with over 20 entering the clinic. He has been an integral member of key leadership teams from target identification through discovery and into early clinical development and led both regional and global medicinal chemistry teams, overseeing budgets and talent strategy. Nick held positions at Janssen Research & Development LLC from 1999 until retirement in 2020. While at Janssen his primary responsibility was to lead medicinal chemistry for the Neuroscience Therapeutic Area ultimately assuming the role of VP and Global Head of Chemistry for Neuroscience. A range of CNS targets were successfully prosecuted and to date ten compounds have advanced to clinical POC including the H3 antagonist JNJ-31001074 (Bavisant) and the orexin-2 antagonist JNJ-42847922 (Seltorexant). Prior to joining Janssen, Nick began his industrial career at Hoechst in the U.K. and subsequently at Schering-Plough where he contributed to several clinical candidates. Nick obtained his B.Sc. (First Class) in chemistry and Ph.D. in synthetic organic chemistry from Heriot-Watt University, Edinburgh, Scotland which was followed by a NATO-Research Fellowship at the University of California, Berkeley. He has one hundred and twenty published papers, several book chapters, and forty-five issued U.S. patents (h-index 45). Nick was elected a Fellow of the Royal Society of Chemistry in 1997.
Steven K. Duddy, Ph.D., Toxicology
Dr. Duddy has over 25 years of toxicology experience in academia and the pharmaceutical industry, and he is currently a founder and Chief Scientific Officer at Integrated Nonclinical Development Solutions Inc. (INDS), a pharmaceutical development consulting firm. Prior to forming INDS in 2007, he spent 11 years at Parke-Davis/Pfizer, where he built a dynamic molecular toxicology group and focused on nonclinical drug safety research and compound development. Dr. Duddy has considerable experience as a drug discovery and development team representative, study director/monitor, author of nonclinical components of regulatory submissions (INDs, NDAs), and in formulating and implementing experimental and regulatory strategies to support development of promising preclinical drug candidates. Additionally, he has experience managing laboratory researchers and resources, solving difficult drug candidate toxicity issues through novel investigative strategies, supporting safety issue resolution for nonclinical drug candidates. Dr. Duddy has published and presented extensively on his research, has contributed to ILSI/HESI working groups on toxicology issues relevant to pharmaceutical development, and he regularly participates on NIH SBIR/STTR grant review panels. Before making the transition to pharmaceutical development, Dr. Duddy held academic research appointments at the W. Alton Jones Cell Science Center in Lake Placid, NY, and in the Department of Pathology, University of North Carolina-Chapel Hill.
Ronald B. Franklin, Ph.D., D.A.B.T., Pharmacokinetics
Dr. Franklin is Chief Scientific Officer and Founder of Franklin ADME Consult, LLC, a consulting company advising pharmaceutical companies on all aspects of ADMET in discovery, development and early clinical studies. Prior to entering big pharma in 1981, he was Assistant Professor in the Department of Pharmacology and Toxicology at The Medical College of Wisconsin where he conducted research into lung toxicity with the aid of an R01 grant from NIH (NHLBI). Subsequently, during his 14 year tenure at Eli Lilly and Co., he worked on predominantly cardiovascular and CNS drugs, including isomazole, quinelorane, flumezapine, olanzapine (Zyprexa) and dapoxetine (sold and later named Priligy). He had a brief spell at Amgen in Boulder, CO and then 7 years at Merck Research Laboratories in Rahway, NJ as Director of Discovery Metabolism and then Director of Development Metabolism involved primarily with PPAR agonists. Dr. Franklin became Senior Director of Drug Metabolism, Pharmacokinetics and Clinical Pharmacology at Array Biopharma, Inc in 2004 where he stayed for 10 years before founding his own company. He has 81 peer-reviewed papers and 4 book chapters. He was on the Editorial Board of Drug Metabolism Disposition for 19 years and he is a Field Editor for the Journal of Pharmacological and Toxicological Methods. He is also on the Editorial Board Current Drug Metabolism and Drug Metabolism Letters. He has been a Diplomate of the American Board of Toxicology since 1981.
Mo Heidaran, Ph.D., Chemistry, Manufacturing & Control (CMC)
Dr. Heidaran is a recognized subject matter expert in biological products, with primary focus on advanced therapies, including cell and gene therapies. With over 35 years of experience in biological drug development, his career has been dedicated to advancing innovation in cell and gene therapy and regenerative medicine across government, industry, and consulting sectors. Dr. Heidaran previously served at the National Cancer Institute (1987–1996) and later at the FDA Center for Biologics Evaluation and Research (2010–2019), where he held multiple leadership roles within OTP and OCBQ. During his tenure at CBER, he served as product and facility reviewer, Acting Team Leader, Acting Branch Chief, and Advisor to the Director of ADRM. While in OCBQ/DMPQ, he developed deep expertise in Current Good Manufacturing Practices (CGMPs) and earned distinction as a Master Reviewer of CGMP at CBER. Since leaving FDA, Dr. Heidaran has advised more than 100 biotechnology companies and federal agencies, including Advanced Research Projects Agency for Health and the National Heart, Lung, and Blood Institute Catalyze Program. At Parexel International, he further expanded his expertise in clinical trial operations, regulatory strategy, and the diverse product portfolio previously regulated under OTAT. Dr. Heidaran is currently Co-owner, CEO, and Chief Regulatory Scientist at Cellx Inc., where he provides end-to-end strategic guidance in biologics development with emphasis on cell and gene therapies. His technical expertise includes manufacturing process development, implementation of process changes, assay development and qualification, validation strategies, and establishment of robust Chemistry, Manufacturing, and Controls (CMC) frameworks to support successful regulatory submissions and scalable commercialization.
James (Jim) Herman, Ph.D., Toxicology
Dr. Herman has more than 34 years’ experience managing the nonclinical development of novel pharmaceuticals from discovery through successful registration. Currently, he is Vice President of Toxicology at Certara USA, leading a group of 9 toxicologists providing strategic, operational, and tactical consulting services to small and large pharmaceutical companies worldwide. Previously, he was President of Integrated Nonclinical Development Solutions, Inc (INDS). Prior to establishing INDS, he spent 20 years at Pfizer and its predecessor in Ann Arbor (Parke‑Davis) in various scientific and managerial roles within the toxicology group. He has significant experience in serving on multidisciplinary drug development teams for small molecules and biologics in several therapeutic areas. He has also served as study director for exploratory and GLP toxicology studies, and is well-versed in study design, execution, and reporting. He also has significant experience in the regulatory toxicology field in all 3 regions (US, EU, and Japan), and has authored and/or critically reviewed over 200 nonclinical portions of IND and NDA/MAA submissions, including sections required for Investigator Brochures and product labeling. He was involved significantly in the development of the first-in-class drugs Cognex® (tacrine), Neurontin® (gabapentin), Rezulin® (troglitazone), Nexletol® (bempedoic acid), Onfi® (clobazam), and the blockbuster drugs Lipitor® (atorvastatin) and Lyrica® (pregabalin). Dr. Herman has also has experience as a nonclinical consultant in the National Institutes of Health’s Blueprint Neurotherapeutics Network.
Christoph A. Kahl, Ph.D., Gene Therapy, Viral Vector Systems, and CMC Strategy
Dr. Kahl is a Subject Matter Expert in cell and gene therapy with over 20 years of experience in the translation of viral vector systems from research to clinical manufacturing. He has held scientific leadership roles at GenVec, Oregon Health & Science University (OHSU), Atara Biotherapeutics, Amgen, and Vir Biotechnology. Dr. Kahl’s technical expertise encompasses the development and scale-up of diverse viral vector platforms, including AAV and lentiviral systems. His work focuses on process and analytical development, optimization for manufacturability, and the design of robust CMC strategies for regulatory submissions. He has contributed to numerous INDs and supported pivotal trials and BLA enablement for Ebvallo™, the first approved allogeneic T-cell immunotherapy. His research background includes the development of AAV-based gene therapies for neurological applications in both mouse and non-human primate models. He is experienced in managing technology transfers and establishing strategic partnerships with domestic and international CDMOs. Dr. Kahl earned his Ph.D. at Indiana University and completed a postdoctoral fellowship at the University of Southern California, focusing on the advancement of gene delivery platforms for clinical application.
Parveen Kaushal, Ph.D. Analytical Development CMC
Dr. Kaushal is a consultant with over 30 years of experience in drug development working with biologics; recombinant proteins, vaccines, mAbs, IG, gene therapy (viral & non-viral) and small molecule pharmaceuticals. Dr. Kaushal is a subject matter expert in analytical development, managing all aspects of physicochemical and biophysical assays, bioassays and In-Vitro cell-based, mechanism of action, potency assays including methods development, qualification, validation, troubleshooting, tech transfers, and technical oversight. Expertise in product characterization, comparability, in-process, cGMP release & stability for bulk and drug product. Dr. Kaushal managed liquid & lyophilized formulation development. Oversaw process development, scale-up and cGMP manufacturing of the drug products. Extensive experience in multiple aspects of operations; analytical, formulation and process development, fill/finish manufacturing, supply chain, product distribution and QC including cGMP compliance. Experience in management, working with CROs and CDMOs, project management, regulatory requirements and FDA submissions (INDs through BLA). Managed multiple concurrent projects, technically oversaw various functions for drug substances and drug products that have advanced from early R&D through pre-clinical and clinical phases (Phase I, II and III) to commercial. Contributed to the advancement of numerous drug products at NABI Biopharmaceuticals, Human Genome Sciences (HGS), UPM Pharmaceuticals, Celsis, PharmAthene, Pfizer and Spark Therapeutics (subsidiary of Roche). Dr. Kaushal holds a B.Sc. (Honors) degree in Chemistry, a Doctorate degree in Physical Organic Chemistry, both from University College, University of London, England (U.K.).
Amy Kimzey, Ph.D., D.A.B.T., Toxicology
Dr. Amy Kimzey is a Senior Managing Scientist with more than 16 years of experience in pharmaceutical discovery and regulatory toxicology. Dr. Kimzey has served as the lead project toxicologist for multiple programs addressing various indications for small-molecule and biologics candidate drugs, and she has advised project teams and colleagues on central nervous system (CNS) drug development. Dr. Kimzey also has experience in discovery toxicology, including identifying and solving toxicology problems during the pre‑candidate stage and identifying potential drug candidates with the highest probability for success in collaboration with multidisciplinary project teams. In addition, Dr. Kimzey is also the lead or co-author of numerous manuscripts, posters, and regulatory submissions, including nine successful IND submissions to FDA and an additional three IND/IND equivalent submissions pending for 2022. She presented a poster titled, “Case Studies Demonstrating the Process and Challenges of Deriving Exposure-Based Limits for Impurities in Pharmaceutical Drug Products Administered Directly to the CNS,” at the American College of Toxicology’s 2021 meeting. Dr. Kimzey has provided toxicology support for multiple CNS drug candidates supported by the NIH HEAL Initiative, including a highly complex pain program for which the small molecule test article is administered via the intrathecal route, as well as an oral small molecule drug candidate to treat pain.
Marc M. Lemaitre, Chemistry, Manufacturing & Control (CMC)
Dr. Lemaitre has over 40 years of experience working in the bio-pharmaceutical industry in various technical and managerial positions at various places. Dr. Lemaitre became independent consultant to provide consulting expertise in CMC and Regulatory Sciences with a focus on assisting clients developing synthetic oligonucleotides [ASOs, siRNAs] for manufacturing, submission and approval. He is knowledgeable in the technical and regulatory documentation requirements for INDs/IMPDs and NDAs/MAAs. Over the years, Dr. Lemaitre has been involved in preparing and/or reviewing more than 20 submissions. He has also made presentations on CMC and regulatory affairs topics as an invited speaker at association meetings and scientific conferences. Dr. Lemaitre is member of the USP expert committee BIO1 [oligonucleotides, peptides and carbohydrates] and of the SAB of the Oligonucleotide Therapeutics Society.
Jason M. LePree, R.Ph., Ph.D., Pharmaceutical Development
Dr. LePree is a pharmaceutical development leader with broad CMC experience spanning preformulation, formulation, analytical sciences, and regulatory authorship from preclinical through late‑stage development. He has nearly 30 years of pharmaceutical development experience, with approximately half in analytical sciences and the other half in preformulation/material characterization and formulation. He is a licensed pharmacist in the State of New Jersey. He has served as a CMC Technical Development Leader, directing cross‑functional teams, global filings (Module 3), and scale‑up/tech transfer across oral, sterile/long‑acting injectable, ophthalmic, and nasal dosage forms. He was the lead formulator and preformulator for A0001—an orphan drug candidate for Friedreich’s Ataxia—advancing lipid‑based IV and oral formulations from preclinical studies into Phase I/II trials and authoring associated IND/IMPD sections. His expertise includes small‑molecule material characterization, lipid‑based delivery for low‑solubility compounds, dissolution/IVIVR, and Quality‑by‑Design. Dr. LePree is Adjunct Professor of Pharmacy at the University of Wisconsin–Madison and an Instructor in Quantitative Analytical Chemistry at Saint Thomas Aquinas College. He previously held technical leadership roles at Boehringer Ingelheim (Animal Health), Viatris (Oyster Point), Ferring, and others, and currently consults via Pyrenees Pharmaceutical CMC Consulting.
Jiunn Lin, Ph.D., Drug Metabolism and Pharmacokinetics
Dr. Lin is an independent consultant who specializes in pharmacokinetics and drug metabolism. He currently provides consultation services for numerous Fortune 500 biotechnology companies in support of their drug discovery and development programs of small molecular compounds as well as large molecular biologics. He was formerly Executive Director at Merck Research Laboratories in charge of drug discovery and preclinical development. During his 26 year career at Merck, Dr. Lin made significant contributions to the FDA approval of 7 blockbuster drugs (Pepcid, Singulair, Fosamax, Crixivan, Trusopt, Cozaar, and Cancidas). Dr. Lin’s vast research experience and interests include dose- and species-dependent pharmacokinetics, in vitro-in vivo drug metabolism, renal handling of drugs, CNS drug delivery, and the role of drug transporters in tissue distribution and CNS transport. Recently, his research interest has focused on the pharmacokinetics of large molecules, including therapeutic monoclonal antibodies, proteins, and peptides. Dr. Lin has published more than 175 peer-reviewed research papers, 28 review articles, 10 book chapters, and 3 patents since 1978.
Nigel J. Liverton, Ph.D., Medicinal Chemistry
Dr. Liverton has 40 years of experience in medicinal chemistry, spanning large pharma, a leadership role at TDI, a translational group formed to advance groundbreaking research at Weill Cornell Medicine, MSK Cancer Center and The Rockefeller University, and contract research organization management. At Merck, he led the medicinal chemistry group that discovered a portfolio of HCV protease inhibitors including two marketed drugs, vaniprevir and grazoprevir, as well as multiple additional candidates that advanced into the clinic. In the CNS field, Dr. Liverton led the NMDA NR2b antagonist medicinal chemistry group where the lead compound advanced into Phase 1b studies for Parkinson's Disease and Phase 2 for treatment resistant depression. He also led groups in cardiovascular and immunology areas that discovered multiple compounds that advanced into IND enabling studies. As VP of chemistry and a member of the leadership team at TDI, he led the medicinal chemistry team, managed RFP's and the evaluation of potential projects, together with strategic oversight of the project portfolio. Successful TDI projects have led to multiple pharma/biotech outlicensing agreements and new company startups. Dr. Liverton is an inventor on 88 issued US patents and author on more than 70 publications.
Paul G. Pearson, Ph.D., Pharmacokinetics
Dr. Pearson is President and Chief Executive Officer of Pearson Pharma Partners, a consulting organization that specializes in pharmacokinetics, drug metabolism and translational science for biopharma and venture capital companies. In prior positions, he served as Global Head and Vice President of Pharmacokinetics and Drug Metabolism at Amgen and as Executive Director of Preclinical Drug Metabolism at Merck Research Laboratories where he was responsible for drug metabolism support for drug discovery and development programs. Dr Pearson has published extensively in the areas of pharmacokinetics, drug metabolism, reactive drug metabolites, drug-drug interactions and drug-induced toxicities, as well as co-edited the Handbook of Drug Metabolism. His research interests are focused on pharmacokinetics and drug metabolism, including the understanding of factors that influence the pharmacokinetics, metabolism, efficacy and safety of novel therapeutic agents in humans. For a period of almost 20 years, Dr Pearson has made major contributions to the approval of important new therapeutics to treat cancer (Camposar, Vectibix), Parkinson’s disease, hematological disorders (Nplate), HIV (Isentress) and fatal fungal infections (Cancidas).
Richard Pfeifer, Ph.D., Toxicology
Dr. Richard Pfeifer is a board-certified toxicologist and seasoned Nonclinical Development strategist with over 25 years of industry experience spanning small molecules, biologics, gene therapies, vaccines, enzyme and mRNA replacement therapies, and medical devices. He has led or contributed to more than 90 programs, including over 35 development track decisions, 30+ IND/CTA submissions, and multiple NDA/BLA/MAA filings, with deep expertise across general, reproductive, genetic, and investigative toxicology, as well as safety pharmacology and PK/ADME. Most recently Vice President of Toxicology, Research and Preclinical Development at AVROBIO, he advanced ex vivo lentiviral gene therapies through global regulatory approvals, major financings, and an asset sale to Novartis. His prior leadership roles at Shire (now Takeda), Wyeth (now Pfizer), and earlier industry positions included overseeing toxicology strategy, guiding due diligence, directing exploratory toxicology groups, and contributing to immunotoxicology initiatives. A former Purdue University faculty member with more than 60 publications, Dr. Pfeifer holds a BS in Biology from Bucknell University and a PhD in Pharmacology from the University of Rochester, followed by postdoctoral fellowships in immunotoxicology at CIIT and NIEHS.
Frank Preugschat, Ph.D., Bioactivity Assay Development
Dr. Preugschat is a senior-level Bioactivity Assay Development SME consultant with Midnight Sun Technologies, LLC. Additionally, he is a paid consultant contributing to enzyme assay development, inhibitor design, chemistry SAR, and translational biomarker development for several bio/pharma companies. He also does pro bono consulting work for academics that work on interesting/challenging enzyme targets or technologies. He has 25 years of work experience at both pharmaceutical and biotechnology companies (both small molecule and antibody therapeutics). His main expertise is in the discovery and optimization of small molecule inhibitors for a wide range of enzymes. He has developed high, medium, and low throughput biochemical assays for the primary isolation and secondary mechanistic characterization of enzyme inhibitors. This work has resulted in numerous publications, issued patents, and the approval of marketed drugs. He is an experienced and talented scientist with the ability to quickly learn new technologies and apply them to scientific problems.
Jeffrey Simard, Ph.D., Assay Development
Dr. Simard is a senior drug discovery leader with over 20 years of experience developing and implementing high-throughput biochemical and cellular assays to support small-molecule discovery in the oncology, immunology, inflammation, and neuroscience therapeutic areas. He has led functional teams responsible for cellular assay development, compound profiling, and automation strategy within both biotechnology and pharmaceutical settings, most recently serving as Senior Director of Cell Pharmacology at a clinical-stage targeted protein degradation company. His expertise centers on the design and execution of integrated screening funnels that span target engagement, pathway modulation, phenotypic profiling, and mechanistic characterization to enable data-driven medicinal chemistry optimization and candidate selection. Dr. Simard has overseen implementation of modular automation and miniaturized 384-well screening platforms, managed large-scale small-molecule screening campaigns, and partnered extensively with CROs and cross-functional discovery teams to accelerate programs from hit identification through IND-enabling studies. A recognized scientific leader and collaborator, he has contributed to multiple clinical assets, authored numerous peer-reviewed publications, and is known for applying innovative assay technologies and strategic insight to advance preclinical drug discovery programs efficiently from early discovery to clinical development.
Robert J. Timko, R.Ph., Ph.D., Pharmaceutical development
Dr. Timko has over 45 years of experience working in the pharmaceutical industry in various technical and managerial positions at Johnson & Johnson and AstraZeneca Pharmaceuticals. Dr. Timko founded RhoTau Pharma Services to provide consulting expertise in the Pharmaceutical and Regulatory Sciences with a focus on assisting clients achieve their product goals of a fast submission and seamless approval, while assuring a cost-effective product and secure supply chain. He has worked with a variety of traditional and novel dosage forms across therapeutic areas and interacted with global Health Authorities on a diverse range of CMC topics. He is well versed in the technical and regulatory documentation requirements for INDs/IMPDs, NDAs/MAAs, and sNDAs/Variations. Dr. Timko is considered a subject matter expert with in-depth knowledge of Quality-by-Design, Process Analytical Technology, and Real Time Release in the current regulatory environment. Over the years, Dr. Timko has been involved in preparing and/or reviewing more than 35 marketing applications either in a pharmaceutical development or regulatory capacity for innovator and generic compounds for a variety of small and large molecule dosage forms, including but not limited to, immediate and extended release solid dosage forms, injectables, and liquids. He has hands-on experience in dosage form development, scale-up to commercial manufacture and production troubleshooting, preparing CMC briefing and regulatory documentation, preparing teams, and leading face-to-face meetings with Health Authorities. Additionally, Dr. Timko has provided technical expertise and served as a subject matter expert witness on formulation development in patent litigation cases. He holds several formulation patents and has written and co-authored numerous articles for peer-reviewed journals and technical publications. He has also made presentations on formulation and process development and regulatory affairs topics as an invited speaker at association meetings and scientific conferences. Dr. Timko is an active member of the American Association of Pharmaceutical Scientists (AAPS), the International Society of Pharmaceutical Engineers (ISPE), the International Academy of Compounding Pharmacists (IACP) and the Pennsylvania Pharmacist Association (PPA). He is a Registered Pharmacist in New Jersey and Pennsylvania.
Kristina Ulrich, Ph.D., M.Sc., E.R.T., Toxicology
Dr. Ulrich is a Senior Managing Scientist with more than 18 years of experience working in or with the pharmaceutical industry, including 15 years as a toxicologist supporting drug discovery and development programs, with a focus on neurology (psychiatry and neurodegeneration) and pain indications. Dr. Ulrich has provided nonclinical study oversight and written regulatory documents for FIH studies and Phase 2 and 3 clinical trials, including five INDs/CTAs. Dr. Ulrich led the nonclinical safety strategy for a neuroscience portfolio in a large pharmaceutical company. This included mentoring project toxicologists, developing nonclinical safety plans, critically reviewing toxicology and safety pharmacology data, and providing data summaries for internal governance interactions and regulatory submissions. Dr. Ulrich is also a former nonclinical assessor at the MHRA in the UK, where she reviewed nonclinical data packages submitted with applications for clinical trials to be conducted in the UK and Europe. Currently, Dr. Ulrich provides scientific and regulatory advice to numerous pharmaceutical companies regarding nonclinical development in support of clinical trials and marketing authorization for a wide range of therapeutic approaches (small molecules, biologics, cell and gene therapies). She specializes in designing and implementing nonclinical safety strategies, as well as health authority interactions, data review, problem solving, and recommendations for further scientific investigations. She has also supported development of an intra-articular gene therapy drug candidate in the NIH HEAL program for pain due to osteoarthritis, including providing guidance on the design of preliminary in vivo safety and biodistribution studies.
Devin Welty, Ph.D., Drug metabolism and pharmacokinetics
Dr. Welty has more than 30 years’ professional experience in Drug Metabolism and Pharmacokinetics and Clinical Pharmacology. His expertise is in the areas of pharmacokinetics, pharmacodynamics and translational medicine of small molecules, biologics and gene therapy. He guides modeling and simulation to support drug discovery and development programs. In his current position as Vice President, Nonclinical and Clinical Pharmacology at Allucent, he is responsible for consulting to sponsors in bringing preclinical assets into early clinical development and supporting early clinical development. Prior to this role, Dr. Welty served as Global Head of DMPK, Shire Pharmaceuticals; Head of Pharmacology and Pharmacokinetics, Forum; Sr. Director, DMPK, Allergan; and Sr. Research Fellow, Neurology, Parke-Davis, Warner-Lambert. He has supported more than 25 worldwide approvals for products in Neurology, Ophthalmology and in Rare Diseases. Dr. Welty holds bachelor’s and doctor’s degrees in Chemistry, Pharmacy and Pharmacokinetics from the University of Florida and the Medical College of Virginia. Most recently, he has served as Chair of the University of Washington, School of Pharmacy, Corporate Advisory Board, and on the Board of Directors, Innovation and Quality Consortium.
Ronald E. White, Ph.D., Pharmacokinetics
Dr. White is President of White Global Pharma Consultants, LLC in Cranbury, New Jersey. He was formerly Distinguished Research Fellow, Pharmaceutical Candidate Optimization at Bristol-Myers Squibb and Vice President at the Schering-Plough Research Institute in charge of all ADME, pharmacokinetics and bioanalytical in both discovery and clinical development. In that position, he developed Noxafil® (posaconazole), PEG-Intron® (pegylated interferon-alpha), Victrellis® (bocepravir), Vorapaxar® (thrombin receptor antagonist), and Zetia® (ezetimibe). Prior to joining industry in 1987, he was Associate Professor of Pharmacology at the University of Connecticut School of Medicine. He is a past member of the Pharmacology Study Section of the National Institutes of Health and of the Drug Metabolism Technical Group of Pharmaceutical Research Manufacturers of America and was Chair of the Gordon Research Conference on Drug Metabolism. He is currently a member of the Editorial Board of the journal Drug Metabolism and Disposition and Adjunct Professor of Chemical Biology in the Rutgers University School of Pharmacy. Dr. White holds a Ph.D. in Organic Chemistry from the University of Wisconsin and completed post-doctoral research in biochemistry at the University of Michigan. He has lectured and published extensively in the areas of drug metabolizing enzymes, pharmacokinetics and drug discovery, and is the holder of five United States patents.
Marcie Wood, Ph.D., Toxicology
Dr. Marcie Wood is a Principal Scientist and Director of ToxStrategies’ Biopharmaceutical/ Pharmaceutical Practice. She is a toxicology consultant with more than 20 years of experience in toxicology and drug discovery and development, including 7 years at the FDA as a pharmacology/toxicology reviewer and supervisor. Through her career, she has gained extensive knowledge of regulatory agency nonclinical expectations for the development of drug products for pulmonary, rheumatology, oncology, and ophthalmology, dermatology, and neurology and pain indications, as well as experience in addressing challenging scientific and regulatory issues (e.g., nonclinical hold deficiencies). In addition, Dr. Wood has experience with unique or complex routes of administration (e.g., intra-articular, intranasal, inhalation, and dermal routes), for both biological and small-molecule products. She also has experience in the design, monitoring, and data interpretation of animal (rodent and non-rodent) toxicology studies (GLP and non-GLP), in developing nonclinical sections (pharmacology, pharmacokinetics, toxicology) of regulatory documents (e.g., preIND, IND, BLA/NDA), and regulatory agency interactions. To date, Dr. Wood has overseen or contributed to the nonclinical aspects of successful regulatory submissions, including nine INDs/CTAs and three NDAs, with another NDA currently under FDA review. She has also supported development of an oral small-molecule drug candidate in the NIH HEAL program, including providing feedback on late-stage developmental toxicology data and recommendations for pharmacokinetic, maximum-tolerated-dose, and dose-range-finding non-GLP toxicology studies.
Gene Williams, Ph.D, Drug Metabolism and Pharmacokinetics
Dr. Gene Williams has more than 25 years of experience in strategic planning and review of drug metabolism and pharmacokinetics-related elements of comprehensive drug development programs (from nonclinical IND enabling studies through NDA/BLA submission and post-marketing studies). Gene worked on over 600 different drugs as a Clinical Pharmacology Team Leader at FDA and over 400 as an FDA Primary Reviewer. Molecule types included non-biologics of all types, peptides, oligonucleotides, glycoprotein mixtures, antibodies and antibody-drug conjugates, liposomes and nanoparticles. The diversity across drug types has continued in his current role as a consultant where he has worked on small molecules through gene therapies. As a post-doctoral fellow at what was then the Neurobiology and Anesthesiology Branch of the National Institute of Dental Research at the NIH, Gene investigated the quantitative effects, i.e., additive-ness, synergy or antagonism, of small molecule drug combinations in a rat analgesia model and opioid-induced pruritus in a primate model. Gene’s doctoral dissertation was on the neurochemistry of feedback control of serotonin release in the rat spinal cord.
External Oversight Committee
The External Oversight Committee helps the BPN program adjust to changing landscapes to continue to provide a unique community scientific resource. Membership on this committee may be supplemented with ad hoc participation of individuals with expertise appropriate for specific programmatic matters within the BPN program and the Division of Translational Research.
Michael J. Detke, M.D., Ph.D.

Dr. Detke is a board-certified psychiatrist and a clinical drug development scientist and executive with research experience since 1986, in biotech/pharma research since 1999, and an adjunct Clinical Professor of Psychiatry at Indiana University School of Medicine since 2000. He has experience across clinical trials of many CNS disorders, and in all phases. He is the Chief Medical Officer at Cortexyme, a clinical-phase biotechnology company developing therapeutics targeting a specific pathogen tied to neurodegeneration, for the treatment of Alzheimer's and other degenerative disorders.
Dr. Detke also served as CMO at Embera NeuroTherapeutics and CoMentis, at which he led clinical development of treatments for addictions, Alzheimer's Disease and Cognitive Impairment Associated with Schizophrenia. As MedAvante CMO, he led a team dedicated to improving signal detection in CNS clinical trials. As Executive Director for Neuroscience Medical Research at Lilly Research Laboratories in Indianapolis, he oversaw all CNS assets in early phase development, including all psychiatric, neurological, and pain indications. Previously at Lilly, he was Senior Medical Director responsible for Phase III development for Cymbalta and Phase IV for Prozac, and a Clinical Research Physician in late-phase development.
Educated at Yale University and the University of Pennsylvania and trained in psychiatry at Harvard Medical School, he has over 70 manuscripts published in peer-reviewed journals and is an active member of scientific organizations such as ACNP, ASCP and SOBP.
Janie S. Merkel, Ph.D.

Dr. Merkel is Director of Research Cores at the Yale University School of Medicine. Through her training and professional experiences in biotechnology companies, she has focused on applying large scale biology technologies to answer biological questions across many fields. She contributes an understanding of biophysical techniques to evaluate proteins’ structures and binding to other molecules. For over 13 years at the Yale Center for Molecular Discovery, she worked with the Center’s team of biologists and chemists in collaboration with faculty to develop biochemical and cellular assays for small molecule screening and to optimize molecules identified in screens. Her specific area of expertise is assay development and result interpretation. During that time, the Center embarked on State-funded collaboration to rapidly translate ideas for novel targets into proof-of-concept data to promote new biotech ventures in Connecticut. The program was open to projects across all therapeutic areas, with a focus on small molecule intervention. She serves as a mentor for a NIDA-funded training course to foster the next generation of entrepreneurs for Substance Use Disorder innovations. Janie received her Ph.D. from Yale University and her bachelor’s degree from Dartmouth College.
Daryl Schoepp, Ph.D.

Dr. Schoepp has over thirty years' experience in the discovery and development of innovative Neuroscience therapeutics in pharmaceutical industry. This includes 20 years at Eli Lilly as a scientist and leader of Neuroscience research department, and 11 years at Merck as the Neuroscience research therapeutic area leader.
As a scientist he has over 200 publications (>19,000 citations of this work) and is an inventor of 15 US patents. He is recognized for having made major contributions in the investigation of the excitatory amino acid neurotransmitter glutamate in disease pathophysiology, pharmacology and therapeutics. His bench and leadership roles have led to the discovery and introduction into patients of over 20 novel first in class agents for psychiatric and neurological diseases. These include the first AMPA/kainate and metabotropic receptor negative and positive modulators (e.g. tezampanel, talampanel, LY354740, LY341595, eglumetad and pomoglumetad) investigated for migraine, pain, cognition, anxiety disorders and schizophrenia. While at Lilly, he was a co-discoverer of the compound LY246736 (alipimovan/Entereg) a first in class peripherally restricted opioid antagonist for post-operative ileus.
At Merck as head of Neuroscience research, his team have built a pipeline of innovative first-class agents for Alzheimer’s disease, Parkinson’s disease, pain/migraine, and schizophrenia. Merck Neuroscience successfully developed and launched the first in class orexin receptor antagonist Suvorexant (Belsomra) for insomnia and created the novel first in class oral CGRP antagonists ubrogepant and atogepant currently in registration trials for migraine treatment and prevention (by Allergan). His group made important contributions to the pioneering trials of the BACE inhibitor verubecestat in prevention trials for Alzheimer’s disease.
Barbara Slusher, Ph.D.

Dr. Slusher is Professor of Neurology, Pharmacology, Psychiatry, Neuroscience, Medicine and Oncology at Johns Hopkins School of Medicine and the Director of Johns Hopkins Drug Discovery. She has published over 250 scientific articles and is the inventor on over 100 patents and patent applications.
Before joining Johns Hopkins, Barbara spent 18 years in the pharmaceutical industry, including several years as Senior Vice President of Research and Translational Development. She has extensive experience in drug discovery through early clinical development and was involved in the successful development, launch and/or post marketing support of four FDA approved medicines.
In 2009 she joined Johns Hopkins to lead the largest drug discovery program on campus with a veteran team of over 25 medicinal chemists, assay developers, pharmacologists, toxicologists, and pharmacokinetics/drug metabolism experts. The team is engaged in identifying novel drug targets arising from the faculty’s research and translating them into new small molecule therapeutics for clinical development.
Since joining JHU, Barbara has co-founded four new companies including Cerecor, Dracen Pharmaceuticals, Adarga, and Lorem Therapeutics amassing over $150M in funding. She also co-founded the first International Consortium of Academic Drug Discovery with over 150 university-led translational centers and 1500 members. Dr. Slusher has served on multiple scientific advisory boards and steering committees including Janssen, Eisai, Inc., Bayer-Wilmer, Bluefield Innovations, Neurofibromatosis Therapeutic Acceleration Program, American Society for Experimental NeuroTherapeutics, Longeviti Neurosolutions, Ashvattha Therapeutics, and the Kennedy Krieger Institute.
Barbara brings broad expertise in drug discovery, pharmacology, toxicology, drug metabolism and pharmacokinetics, and animal disease modeling to the BPN.
Terence A. Kelly, Ph.D.

Dr. Kelly is a consultant with over 30 years of experience in the pharmaceutical industry in the US and Europe. After finishing postdoctoral studies at Yale in 1990, he worked as a medicinal chemist at Boehringer Ingelheim on projects in the areas of Immunology, Virology, CNS and Cardiovascular diseases. From 2002 to 2009 he led BI's medicinal chemistry department in the US, which included 145 scientists in the high throughput screening, computational chemistry, structural biology, combinatorial chemistry and medicinal chemistry groups. He joined CoMentis in 2010 as Chief Scientific Officer and became the CEO in November 2011. From 2019 to 2023, he was the CEO of Perception Neuroscience and led that company through its Phase 2a trial in Treatment Resistant Depression. He currently consults for several Biotech and VC companies on topics ranging from discovery to early clinical strategies.
Terence A. Kelly, Ph.D.